 Cuore Sano
Cuore Sano
LA SINDROME DEL Q-T CORTO,
SHORT Q-T SYNDROME (SQTS)
============================
Da alcuni decenni è noto che l'intervallo Q-T lungo (> 0,460 sec) si associa a un elevato ri-schio di morte improvvisa. Al contrario finora non era noto quale fosse il significato clinico di un in-tervallo Q-T < 0,300 sec. Solo di recente è stato osservato che anche il Q-T breve si caratterizza per una instabilità elettrica ventricolare e per un elevato rischio di morte aritmica.Trattasi in sintesi di una nuova entità nosografica, detta "Sindrome del Q-T corto" (Short Q-T Syndrome, SQTS).

E' una sindrome rara rispetto alla ben più nota sindrome del Q-T lungo, ma anch’essa causa di morte improvvisa nella popolazione. Si tratta di una malattia genetica del sistema elettrico del cuore che si trasmette come carattere autosomico dominante.
In sintesi la sindrome si caratterizza per:
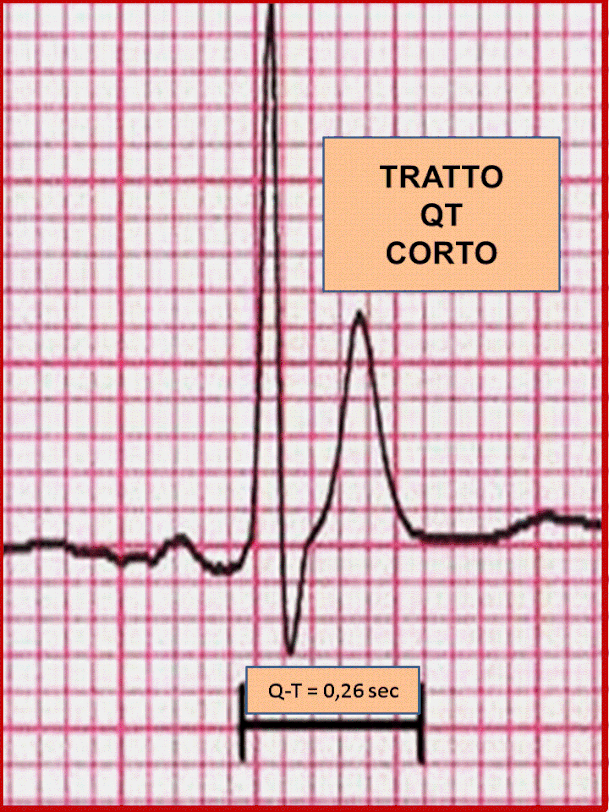
.intervallo Q-T breve all'
ECG (≤ 0, 300 sec) che non si modifica in maniera significativa con il ritmo cardiaco;
.onde T alte e
appuntite;
.cuore strutturalmente
normale.
La sindrome è stata descritta per la prima volta nel 2003, dal prof Fiorenzo Gaita di Torino, il qua-le in un bambino di 8 mesi, rianimato per arresto cardiaco aveva osservato all'ECG un intervallo Q-T assai corto.

Dopo questa osservazione, Gaita raccogliendo l' anamnesi familiare del bambino, osservò che la madre soffriva di episodi sincopali che fino a quel momento erano stati attribuiti ad una sincope va-so-vagale.L'elettrocardiogramma della madre e di uno zio materno però rivelavano le stesse carat-teristiche osservate al bambino,cioè un tratto Q-T corto,intorno a 0,250-0,260 sec. Oltre a questo rilievo elettrocardiografico nell'arco di 4 genera-zioni, in quella famiglia si erano verificati 8 casi di morte improvvisa.
Di alcuni di questi pazienti esistevano i reperti autoptici che avevano mostrato un quadro di normalità dell'appa-rato cardiocircolatorio. Con l'esecuzione di esami stru-mentali anche l'apparato cardiocircolatorio della madre e dello zio risultavano normali. Gaita, successivamente da esperto elettrofisiologo,effettuò uno studio elettrofisio-logico del cuore e dimostrò che, attraverso l'erogazione di stimoli elettrici prematuri nell'arco di tutto il ciclo car-diaco,era possibile la trasmissione dello stimolo ai ventri-coli dando luogo ad una crisi di fibrillazione ventricolare.
MA PERCHE' FINORA NON SONO STATI DESCRITTI CASI DI SINDROME DEL Q-T CORTO?
E' verosimile che come accade per la sindrome del Q-T lungo, anche i pazienti con Q-T Corto muoiano in età pe-diatrica prima dell' esecuzione di un ECG e quindi molti di essi passavano inosservati. Da quando, per l' intui-zione del cardiologo torinese, la sindrome è stata de-scritta, in letteratura via via ne vengono diagnosticati alcuni casi; però bisogna precisare che i casi con sin-drome del Q-T corto sono fortunatamente abbastanza rari. Attualmente infatti sono solo poche decine i casi ri-portati a livello mondiale.
QUALE TRATTAMENTO
L'alta incidenza di morte improvvisa in questi pa- zienti ha reso il defibrillatore impiantabile la scelta migliore per la prevenzione della morte improv- visa, sin dalla prima osservazione.
Ciononostante a causa della giovane età dei pa- zienti e dell'elevata incidenza di complicanze è e-mersa la necessità di trovare una terapia farma- cologica in grado di agire sulle proprietà elettro- fisiogiche delle cellule cardiache. Sono stati testati numerosi farmaci come sotalolo, ibutilide, flecai-nide, disopiramide, amiodarone, dofetilide, nife-kalant, ma gli effetti elettrofisiologici e clinici sono risultati controversi.
In conclusione secondo Gaita per i soggetti con arresto cardiaco rianimato o con sincope aritmica, la strategia terapeutica migliore è rappresentata dall'impianto di un defibrillatore automatico im- piantabile.Per i soggetti asintomatici,in assenza di elementi per la stratificazione del rischio arit- mico, si può proporre un trattamento profilattico con idrochinidina e l'impianto di un dispositivo loop recorder sottocutaneo per monitorare l'even-tuale insorgenza di aritmie pericolose. Nei pazien-ti in cui si rilevano aritmie sostenute l'impianto di un defibrillatore rimane ovviamente mandatorio.
Un articolo del 2013 ha affrontato gli effetti elettrofisiolo-gici del trattamento β-bloccante sui canali del potassio alterati da mutazioni geniche alla base della sindrome del Q-T breve. Sono state analizzate 2 mutazioni geni-che che comportano un “guadagno di funzione” delle cor-renti in uscita attraverso i canali del potassio responsabi-li della fase di ripolarizzazione (IKr e IKs), mutazioni che vanno sotto la sigla N588K-KCNH2 e V307L-KCNQ1 e che condizionano le sindromi SQT1 e SQT2, nelle quali il trattamento β-bloccante con carvedilolo e con metoprolo-lo è stato testato in clinica. Si tratta di uno studio in vitro su cellule isolate. In breve gli autori hanno osservato che il carvedilolo e il metoprololo differiscono signififativa-mente nella loro capacità inibitoria sui canali del potas-sio, essendo il metoprololo molto più potente del carve-dilolo.
Poichè lo studio è stato effettuato in laboratorio i risultati sono di difficile applicazione immediata in campo clinico. Lo studio tuttavia sottolinea come gli effetti del tratta-mento β-bloccante possano differire in modo significativo da una molecola all’altra. J Cardiovasc Electrophysiol maggio 2013.
INDICE ANALITICO





