 Cuore Sano
Cuore Sano
LA SINDROME DI
BRUGADA
(Brugada Syndrome)
La sindrome di Brugada è una entità clinica de-scritta nel 1992 dai fratelli Brugada,Pedro e Josef. E' secondaria ad uno squilibrio tra le correnti ioni-che entranti e uscenti dalle cellule cardiache. In genere è responsabile una ridotta funzione dei ca-nali che conducono la corrente entrante del sodio.

La presenza di una corrente uscente di potassio (Ito),che è particolarmente rappresentata a livello del tratto di ef-flusso del ventricolo destro e che non viene controbilan-ciata da quella del sodio si rende manifesta con un parti-colare aspetto ECGrafico in questa particolare sede. Il ri-schio aritmico deriva dalla eterogeneità nella polarizzazi-one di aree miocardiche adiacenti,dovuta allo squilibrio tra le correnti ripolarizzanti e depolarizzanti.Questo favo-risce fenomeni di rientro dovuti alla presenza di extrasi-stoli ventricolari precoci con il fenomeno R su T,che pos-sono innescare la fibrillazione ventricolare e l’arresto car-diaco. Tutt'ora l'anomalia è responsabile del 4% di tutte le cause di morte cardiaca improvvisa.In Europa la Sin-drome di Brugada ha una diffusione relativa bassa; in Ita-lia si stima che ogni 10.000 nati, nascano da 0,4 a 1,7 bimbi malati, con una prevalenza dello 0,7-2,5%. Questo dato sale al 6% in Francia, all'11% nelle nazioni del Me-dio Oriente e arriva al 12% in Thailandia. I maschi sono più spesso sintomatici rispetto alle femmine,forse per l'in-fluenza degli ormoni sessuali sulle aritmie cardiache e/o canali ionici e una diversa distribuzione dei canali ionici attraverso il cuore in maschi versus femmine.In molti casi alla sindrome di Brugada si associano altre patologie co-me la displasia aritmogena del ventricolo destro o la sin-drome del QT lungo e del QT corto,condizioni che hanno un effetto moltiplicativo sul pericolo di morte improvvisa.
La sindrome di Brugada riconosce una origine genetica con trasmissione autosomica dominante a penetranza in-completa e si associa a più di 250 mutazioni in 11 geni diversi.La prima mutazione che è stata associata alla sin-drome è una alterazione nel gene che codifica per la proteina che costituisce il canale del sodio. Allo stato at- tuale una specifica mutazione genetica viene identificata solo nel 18-30 % dei pazienti con sindrome di Brugada, ma vengono descritte sempre nuove mutazioni a carico di questo e di altri geni.
L'alterazione dei canali ionici si accompagna ad alte-razioni elettrocardiografiche caratteristiche e ad aritmie ventricolari maligne causa di sincopi e di morte improv-visa. L'incidenza di eventi fatali è più elevata nei maschi nella terza e quarta decade di vita.
Le caratteristiche elettrocardiografiche comprendono un sopraslivellamento del tratto ST nelle derivazioni precor-diali di destra (V1-V2-V3) cioè nelle derivazioni che regi-strano l'attività elettrica del tratto di efflusso del ventricolo destro.Le modificazioni ECGrafiche non sono costanti, nel senso che alcuni pazienti possono attraversare lun-ghi periodi durante i quali l’ECGramma risulta essere normale.
Pedro Brugada e Josep Brugada sulla rivista Journal of the American College of Cardiology Volume 20, Issue 6, 15 November 1992,Pages 1391-1396"Right bundle bran-ch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome: A multicenter report".
Così descrissero la sindrome che prese il loro no-me.
"Results.The ECG during sinus rhythm showed right bun-dle branch block, normal QT interval and persistent ST segment elevation in precordial leads V1to V2–V3, not explainable by electrolyte disturbances,ischemia or struc-tural heart disease. No histologic abnormalities were found in the four patients in whom ventricular biopsies were performed.The arrhythmia leading to (aborted) sud-den death was a rapid polymorphic ventricular tachy-cardia initiating after a short coupled ventricular extrasy-stole. A similar arrhythmia was initiated by two to three ventricular extrastimuli in four of the seven patients stu-died by programmed electrical stimulation. Four patients had a prolonged HV interval during sinus rhythm. One patient receiving amiodarone died suddenly during im-plantation of a demand ventricular pacemaker. The ar-rhythmia of two patients was controlled with a beta-adrenergic blocking agent. Four patients received an implantable defibrillator that was subsequently used by one of them, and all four are alive. The remaining patient received a demand ventricular pacemaker and his arrhythmia is controlled with amiodarone and diphenylhy-dantoin.Conclusions. Common clinical and ECG features define a distinct syndrome in this group of patients. Its causes remain unknown".

Per poter definire la sindrome di Brugada oltre all' ECG è necessario conoscere l'anamnesi,in partico-lare se sono presenti episodi di sincope, aritmie ventricolari, una storia familiare di morte improv-visa in congiunti di età < 45 anni e una inducibi-lità di tachicardia ventricolare con la stimolazione programmata. Le modificazioni elettrocardiografi-che sono evidenziabili con la somministrazione degli antiaritmici di classe Ia, Ic e III. Il test farma-cologico è basato principalmente sull’uso della flecainide; questo farmaco antiaritmico è in grado di far emergere in presenza della sindrome di Brugada il profilo elettrocar-diografico caratteristico.
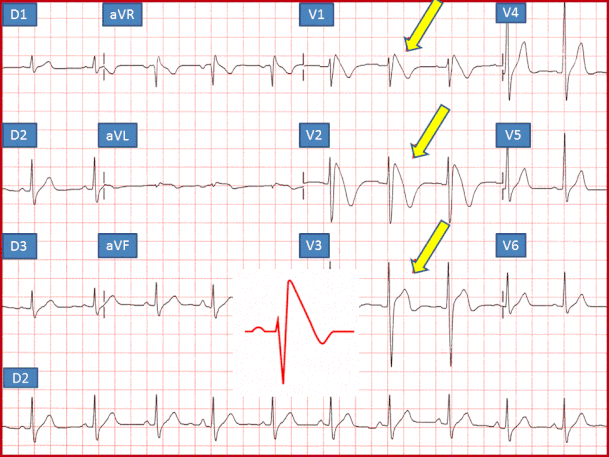
Sono stati riconosciuti 3 tipi di patterns elettrocar-diografici, talora riscontrabili nello stesso paziente in tempi diversi:
1.tipo 1 con sopraslivellamento ST > 2 mm convesso ad andamento discendente, seguito da onda T negativa;
2.tipo 2 caratterizzato da onde J > 2 mm ed ST sopra-slivellato >1 mm, a sella, piatto o ascendente, seguito da onda T positiva;
3.tipo 3 caratterizzato da sopraslivellamento del tratto ST < 1 mm e sopraelevamento del punto J < 1 mm, in pre-senza di un’onda T positiva.Il tipo 3 non è infrequente nei soggetti sani; è abitualmente ritenuto aspecifico, se non converte spontaneamente in un tipo 1.

E' diagnostico solo il tipo 1 che ha una prevalenza nella popolazione generale intorno allo 0,1%.
Secondo alcuni Autori, la diagnosi differenziale tra Bloc- co incompleto di branca destra, in cui coesista un so- praslivellamento del tratto ST e la sindrome di Brugada tipo 2, si basa sulla ripidità della fase discendente dell' onda R' in V1-V2. L'angolo beta tra le rette che prolunga- no l'onda S e la R1 è stretto nel caso del blocco incom-pleto di branca destra e largo quando trattasi di sindro-me di Brugada di tipo 2 (Chevalier S et Al, JACC 2011; 58: 2290-8).

Per la diagnosi vengono impiegati alcuni test con farmaci bloccanti i canali del sodio.Usualmente vengono somministrati flecainide,ajmalina,procai-namide ev. La somministrazione di farmaci bloc-canti i canali del sodio in questi pazienti, accen-tua le alterazioni elettrocardiografiche presenti al-l’elettrocardiogramma di base o le slatentizza nei casi in cui non sono presenti basalmente.
La figura sottostante tratta da un lavoro di Wilde e Altri mostra l'elettrocardiogramma di un giovane di 35 anni, che dopo un arresto cardiaco, era stato rianimato con successo. Da sinistra a destra si osservano 2 strisce elettrocardiografiche e altre ad 1, 2, 3, 4 e a 5 minuti dopo l'infusione venosa di 50 mg di ajmalina.
L'ECG nelle derivazioni V1-V2-V3 della striscia di sini-stra mostra il cosiddetto saddle-back (a sella) pattern, che si caratterizza per un sopraslivellamento del tratto ST di almeno 1 mm e con un'onda T bifasica,cioè un pat-tern tipico della sindrome di Brugada tipo 2, nelle strisce successive comincia a delinerasi il cosiddetto pattern "coved", caratteristico della sindrome di Brugada tipo 1.
La presenza di un ECG tipo 1 e di sincope sono legati sicuramente ad una alta incidenza di arit-mie ventricolari maligne.In questi casi attualmen-te, l'uso del defbrillatore, è la sola terapia possibi-le e ha un'indicazione di classe IA.

Oltre al sopraslivellamento del tratto ST, il quadro ECG può comprendere anche:
.blocco A-V di I grado; è stato riportato che il ritardo di conduzione è sottohissiano e che l’intervallo H-V è pro-lungato nella maggior parte dei pazienti;
.disturbi di conduzione intraventricolare; sono stati de-scritti casi di blocco di branca destra isolato o asso-ciato a emiblocco anteriore sinistro. Nonostante sia sta-to ripe-tutamente affermato, in base alla presenza di una posi-tività terminale del QRS in V1 V2, che il blocco di branca destra sia un carattere pressocchè obbligatorio del feno-meno di Brugada, in realtà non si tratta di un vero blocco di branca né di un ritardo della conduzione intraventrico-lare. La positività terminale del QRS nelle precordiali destre non rappresenta un onda R’,espressione di attiva-zione ritardata del ventricolo destro,ma una deflessione che si genera per il crearsi di un gradiente elettrico fra l’ epicardio e l’endocardio del tratto d'efflusso del ventricolo destro.
Nei pazienti con sindrome di Brugada le pericolo-se aritmie insorgono tipicamente in condizioni di ipertono vagale e quindi durante il riposo nottur- no, durante la fase post prandiale, etc.
A tutt'oggi, non esiste una terapia specifica, ma solo misure di prevenzione che comprendono il controllo di eventuali alterazioni dismetaboliche, degli stati febbrili, l'astensione dall’assunzione di alcuni farmaci e l’impianto di ICD (Impiantable Cardioverter Defibrillator).
Quando devono essere poste le indicazioni all'impianto del Impiantable Cardioverter Defibrillator ?
IN BASE ALLA VALUTAZIONE POLIPARAMETRICA E' POSSIBILE L'ANALISI DEL RISCHIO DI MORTE IM-PROVVISA.
Al centro del sistema c'è il pattern tipo 1 (basale o indotto dai farmaci). Intorno ad esso ruotano 3 fattori di rischio principali:
.sincope,
.familiarità per morte
improvvisa,
.studio elettrofisiologico positivo (vedi
ta-bella sottostante).
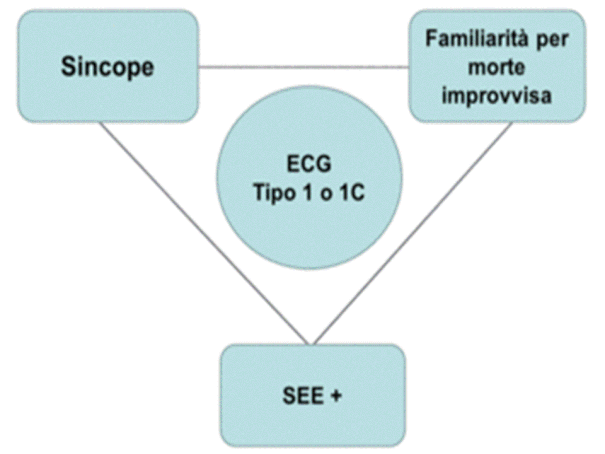
I soggetti a rischio sono quelli con almeno 2 dei citati fat- tori di rischio;nei soggetti di tipo 1 spontaneo e con 2 fat-tori di rischio l'incidenza di eventi maggiori a 3 anni è del 30%,mentre nei pazienti con tipo 1 indotto da farmaci il rischio è < 2% e limitato solo ai soggetti con 2 fattori di rischio (Delise P et Al, EHJ, 2011;99:53-7).
In sintesi non tutti i pazienti con segni ECG tipo Brugada sono ad elevato rischio di morte improvvisa. Soltanto nei casi di positività del test per aritmie ventricolari pericolo-se e nei casi definiti ad elevato rischio, viene proposto il posizionamento di un defibrillatore impiantabile (ICD).
QUALI COMPORTAMENTI BISOGNA OSSERVA-RE IN CASO DI SINDROME DI BRUGADA?
.In caso di febbre bisogna assumere prima possibile antipiretici per abbassare la temperatura corporea;
.Evitare assunzione di droghe e di alcool.
.Evitare pasti molto abbondanti in quanto l'iperattiva-zione vagale da distensione gastrica è pericolosa;
.Evitare l’assunzione di alcuni farmaci assunti per altre patologie come:
1)Farmaci antiaritmici: Flecainide,Propafenone, Aimalina, Procainamide, Disopiramide;
2)Farmaci bloccanti i canali del calcio: Verapamil, Nifedi-pina, Diltiazem;
3) Betabloccanti come Propanololo, Nadololo;
4) Nitrati: Isosorbide Dinitrato, Nitroglicerina;
5)Antidepressivi Triciclici: Amitriptilina, Nortriptilina, Desi-pramine, Clomipramide;
6) Antidepressivi Tetraciclici: Maprotiline;
7) Fenotiazinici: Perfenazine, Cyamemazine;
8)Inibitori selettivi del reuptake della serotonina: Fluoxe-tina;
9) Anestetici/analgesici: Propofol, Bupivacaina, Procaina.
Al 12° Congresso Nazionale AIAC, 12-15 marzo 2015 è stato presentato uno studio per valutare l’outcome a lun-go termine dei pazienti affetti da sindrome di Brugada e sottoposti ad impianto di ICD. La diagnosi è stata for-mulata dopo un episodio di arresto cardiaco resuscitato, un episodio sincopale,durante screening familiare di pa-zienti affetti,occasionalmente durante l’esecuzione di esa mi di routine.Lo studio monocentrico,retrospettivo,ha in-cluso 34 pazienti arruolati dal luglio 1997 al dicembre 2013, che presentavano un pattern ECG tipo I spontaneo o indotto dal test alla flecainide. Sono stati considerati i seguenti dati:età al momento della diagnosi, sesso, mo-dalità della diagnosi, pattern ECG, storia familiare di mor-te improvvisa (<45 anni), risultato del test alla flecainide e dell’eventuale studio elettrofisiologico intracavitario, sintomatologia,follow-up ambulatoriale dall’impianto dell’ ICD sino a maggio 2014. Sono stati considerati sinto-matici i pazienti con storia di sincope, tachicardia ven-tricolare sostenuta documentata,arresto cardiocircolato-rio resuscitato.
Nel 79% dei pazienti è stato eseguito uno studio elettro-fisiologico intracavitario.
I pazienti sono stati suddivisi in 3 gruppi:
A) sopravvissuti ad arresto cardiaco (3 pz),
B) sincope (12 pz),
C) asintomatici (19 pz).
Durante un follow-up medio di 105±49 mesi (intervallo compreso tra 16 e 202 mesi) non si è verificato nessun decesso.Uno shock appropriato si è verificato in 6 pa-zienti (18%) della popolazione totale, significativamente (p=0,02) maggiore nel gruppo A [67% gruppo A; 25% gruppo B; 5% gruppo C], con un’ incidenza annuale dello 0,23% ± 0,08%. Il numero medio di episodi per ciascun paziente è stato 2±0.5. Il tempo intercorso fra l’ impianto del device e il primo shock è risultato in media pari a 22±23 mesi, con nessuna differenza significativa nei tre gruppi. Dall’analisi univariata è stato dimostrato che la presenza di ECG tipo I spontaneo,aritmie inducibili,fami- liarità per morte improvvisa e anamnesi di tachicardie ventricolari sostenute e non sostenute, non differiscono significativamente nei pazienti con shock appropriato delle 3 popolazioni. Diversamente, si è osservato che i pazienti con shock appropriato appartenenti al gruppo A, avevano un’incidenza significativamente maggiore di fi-brillazione atriale (p=0,02).L’incidenza di interventi inap-propriati durante il periodo di follow-up si è verificata in 6 pazienti (18%) con una percentuale annua di 0,37% ± 0,31%. Il numero medio di episodi per ciascun paziente è stato 3±2,4. Tra le complicanze si sono verificate con la stessa incidenza dislocamento di elettrocatetere ventri-colare(3%),versamento pericardico(3%),estrazione trans-venosa di elettrocateteri (3%),senza alcuna differenza si-gnificativa nei 3 gruppi. In questo studio una storia di ar-resto cardiaco resuscitato e/o di sincope sono risultati predittivi di eventi aritmici.L’incidenza di fibrillazione atriale,nella popolazione che ha presentato interventi inappropriati del defibrillatore, è risultata significativa-mente superiore nei pazienti sopravvissuti ad un arresto cardiaco,dato quest’ ultimo compatibile con un’espressi-vità fenotipica peggiore della malattia.
Di recente per la cura della sindrome di Brugada è stata proposta l'ablazione.
Ma di cosa si tratta?
Il dottor Carlo Pappone,elettrofisiologo italiano,ha portato avanti la tecnica dell'ablazione sperimen-tandola prima su 14 pazienti.

Come asserisce Pappone l'intervento viene eseguito in anestesia generale e prevede una degenza ospedaliera di pochi giorni (3-4 giorni). In particolare Pappone dice che mediante la puntura della parte inferiore dello sterno viene introdotto un sondino che viene spinto fino a raggiungere l'epicardio e ne rileva i segnali elettrici. In questo modo viene ricostruita l'anatomia e l'attività elet-trica del cuore, individuando con precisione il punto esatto del ventricolo destro in cui si esprime la malattia.In base all'estensione della zona compromessa che può essere estesa da alcuni cm2 a molti cm2, fino ad arrivare a 1/3 dell'intero ventricolo,saranno necessarie da 5 a 30 applicazioni di energia a radiofrequenza.In sintesi se finora l'unica terapia possibile per i pazienti con Sindro-me di Brugada era l'impianto di un defibrillatore, secondo Pappone il trattamento ablativo ha come obbiettivo quello di trattare quella quota di pazienti affetti dalla Sindrome di Brugada che subiscono ripetuti "shock" terapeutici erogati dal defibrillatore per interrompere le aritmie ventricolari maligne. Nello studio di Pappone,14 pazienti di età media 39 anni affetti da sindrome di Brugada, già portatori di ICD, veniva effettuata la mappatura epicardica per identificare le aree da sottoporre ad ablazione con radio-frequenze prima e dopo somministrazione (2mg/Kg/10min) di flecainide. Venivano individuate, sulla parete anteriore libera e sul tratto di efflusso del ventricolo destro,aree a basso vol-taggio (<1,5 mV) la cui superficie aumentava dopo fle-cainide da 17,6 cm2 (range 12,1 a 24,2 cm2) a 28,5 cm2 (range 21,6-30,2 cm2) (p = 0,001). Dopo 23,8 minuti di radiofrequenza, si riscontrava la sostituzione delle aree a basso voltaggio da aree cicatriziali (<0,5mV).
L’eliminazione del substrato aritmico determinava anche la normalizzazione delle anomalie elettrodiografiche di superficie che persistevano durante il follow-up (anche dopo stimolo con flecainide) senza induzione di aritmie ventricolari maggiori e complicanze.Dopo i 14 casi pub-blicati da Pappone,altri 8 pazienti sono stati operati con successo, nel Ravennate al Maria Cecilia Hospital di Cotignola, dove il dottor Pappone ha lavorato prima di arrivare a San Donato Milanese. Pappone riguardo ai suoi pazienti, sottoposti all'intervento riferisce che erano portatori di defibrillatore che non è stato espiantato a nessuno, ciononostante precisa Pappone, che non è escluso che in futuro, se i risultati perdurassero a lungo termine, questa strategia potrebbe rappresentare un' ipotesi terapeutica da preferire. Pertanto mancando i mo-tivi che giustifichino l'impianto del defibrillatore, secondo Pappone è possibile che l'ablazione diventi non solo complementare,ma alternativa al defibrillatore,per cui se il risultato persisterà nel corso degli anni, il defibrilla-tore,nei pazienti già impiantati, potrebbe essere espian-tato o non sostituito quando la batteria sarebbe scarica.
Al Congresso Nazionale della Società Italiana di Car-diologia, tenutosi a Roma 11-14/12/2015, Pedro Bru-gada, Professore di Medicina presso l'Università di Bar-cellona,nell’ambito di una lezione magistrale,ha com-mentato i risultati dello studio condotto dal dottor Carlo Pappone, nel quale viene descritta la metodica in grado di annullare i segni clinici della sindrome di Brugada. In particolare il Prof Pedro Brugada ha ribadito testualmen-te:"Avendo pubblicato la prima relazione sulla sindrome di Brugada insieme a mio fratello Joseph nel 1992, mi sento autorizzato a rivolgere alcune critiche importanti circa le dichiarazioni rilasciate dal Dottor Pappone.

"Mi sento in dovere di farlo a causa delle aspettative sbagliate che queste dichiarazioni hanno creato tra i pa-zienti, spinti dalla vana speranza di poter definitivamen-te “guarire” da questa malattia.Il Dottor Pappone riferisce di presentare una "nuova idea" e un "nuovo trattamento curativo per la sindrome di Brugada" che, in realtà, è già stata descritta dal collega Koonlawee Nademanee dell' Università della California, circa 4 anni fa. In realtà, non c’è nulla di nuovo; Nademanee aveva già dimostrato l’uti-lità delle procedure di ablazione in tali pazienti nello stu-dio Prevention of ventricular fibrillation episodes in Bru-gada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium (Nademanee K et all; Circulation 2011 Mar 29;123 (12): 1270-9). Il punto nodale,pertanto,non si riferisce al trattamento stesso (pe-raltro,già dimostratosi efficace),ma a chi dovrebbe esse-re rivolto questo trattamento, e se questo può rappresen-tare una valida alternativa all’impianto del defibrillatore (ICD). Con migliaia di pazienti con sindrome di Brugada in tutto il mondo trattati con un ICD, nessuna pubbli-cazione ha messo in discussione il ruolo fondamentale di questa terapia per prevenire la morte cardiaca improv-visa. Questi pazienti, in genere sono giovani (età media 40 anni) e possono avere una vita normale, nonostante l’ impianto del dispositivo. Dopo l'impianto, non sono poste limitazioni per quanto riguarda lo sport o altre attività, in quanto essi sono costantemente protetti dall’ICD. Ri-guardo allo studio di Pappone, trattasi di un piccolo nu-mero di individui, cioè 14 pazienti che erano già impian-tati con un Impiantable Cardioverter Defibrillator. Il trat-tamento ablativo ha, quindi,come solo obbiettivo quello di trattare la quota di pazienti affetti dalla sindrome che subiscono ripetuti ‘shock’ appropriati erogati dal defibril-latore. L’ablazione quindi può essere presa in considera-zione in casi estremi ma, di certo, non ‘guarisce’ una ma-lattia genetica”. Congresso SIC Roma 2015.
Nel 2016, in un update sulla sindrome di Brugada, pub-blicato a cura dell'Associazione Italiana di Aritmologia e Cardiostimolazione, il Prof Pedro Brugada,in merito all’a-blazione del substrato si è mostrato favorevole alla tec-nica,che anche nel suo centro, viene eseguita già da alcuni anni,in contemporanea quasi con Nademanee. Tuttavia,ha sottolineato che il campo di applicazione se-condo Lui si stringe ai pazienti con aritmie cliniche o storm aritmici. Non ritiene invece che possa avere un ruolo di prevenzione in pazienti che non hanno aritmie e ha espresso dubbi sulla durata del follow-up in quanto spesso gli eventi aritmici si verifichino a distanza di pa-recchi anni.
Se l’ablazione epicardica potrà sostituire l’impianto del defibrillatore nei pazienti a rischio,Brugada ha confermato i dubbi già espressi per il follow-up.
Riguardo al sesso, chi è più colpito dalla sin-drome di Brugada?
La Sindrome di Brugada è una patologia caratterizzata da un aumentato rischio di morte cardiaca improvvisa. Come è stato detto la diagnosi si basa sul pattern Elet-trocardiografico caratterizzato da un sopralivellamento del tratto ST nelle precordiali destre.
Interessa principalmente giovani uomini adulti ed è infatti molto meno frequente nelle donne, le quali sono stati-sticamente meno esposte al rischio di eventi cardiova-scolari. L'età pediatrica è coinvolta raramente. Colpisce soprattutto la razza asiatica e i suoi sintomi si manife-stano con maggior frequenza quando i pazienti si trova-no in uno stato febbrile, scaturito da qualunque altra con-dizione patologica, come ad esempio un'influenza.
Quindi la patologia è 8-10 volte più frequente negli uo-mini rispetto alle donne ed inoltre generalmente il sesso maschile presenta una prognosi peggiore.Sono diversi gli studi che hanno confrontato i due sessi in termini di prognosi e presentazione clinica.A tal proposito,Bertho-me et al hanno pubblicato su Heart Rhythm uno studio con lo scopo di descrivere le caratteristiche cliniche ed i fattori di rischio aritmici nelle donne affette da Sindrome di Brugada.Le donne con Sindrome di Brugada hanno una espressione clinica della malattia meno aggressiva, presentano meno frequentemente un pattern spontaneo ECG-grafico tipo 1 e sono più frequentemente asintoma-tiche.I fattori di rischio identificati nello studio sono un’ anamnesi positiva per morte cardiaca improvvisa o sin-cope, un QRS frammentato ed i disturbi di conduzione.
Gli ormoni sessuali, inoltre, modulano i canali ionici di-stribuiti diversamente nei due sessi e potrebbero essere considerati responsabili della diversa espressione della patologia nel sesso maschile e femminile nelle diverse fasce di età. La gestione delle donne affette da Sindrome di Brugada dovrebbe essere influenzata,dunque,dai risul-tati provenienti da questi studi.
1.Hiroshi Morita.Gender difference in Brugada syndrome: Mirror images of long QT syndrome?Heart Rhythm 2018; DOI:10.1016/j.hrthm. 2018.09.004
2.Berthome P,Tixier R,Briand J,et al.Clinical Presentation and follow up of women affected by Bruga-da Syndrome.Heart Rhythm 2019;16(2): 260-267.
Sin dalle prime descrizioni, risalenti agli inizi degli anni ’90, la sindrome di Brugada è stata considerata esclusivamente un disturbo dell''impianto elettrico del cuore, in apparente assenza di alterazioni del muscolo cardiaco. Una recente ricerca ha dimostrato che impor-tanti alterazioni del muscolo cardiaco sono alla base del-le alterazioni elettriche e delle aritmie fatali, sono presen-ti nella maggior parte dei pazienti.
Lo studio pubblicato sul Journal of the American College of Cardiology e condotto presso la Fondazione Policlini-co Universitario A. Gemelli IRCCS di Roma, ha dimostra-to che nel 75% dei casi di sindrome di Brugada, alla ba-se delle aritmie cardiache tipiche della patologia c’è un’ anomalia del muscolo cardiaco stesso e uno stato in-fiammatorio anomalo.La scoperta permetterà di predire quali dei pazienti con la sindrome sono a rischio di arit-mie e di morte cardiaca improvvisa, soprattutto in sog-getti giovani altrimenti considerati completamente sani.
Lo studio si è basato sul prelievo di campioni di muscolo cardiaco e sulla mappatura elettrica del cuore in pazienti che sono stati preliminarmente sottoposti ad esame elet-trocardiografico diagnostico per Sindrome di Brugada, evidenziando delle aree con anomalie sul fronte del fun-zionamento elettrico. E’ stato poi eseguito lo screening genetico dei pazienti.È in particolare dall’esame al micro-scopio dei campioni biologici prelevati che è emerso che in oltre il 75% dei casi è presente un’ infiammazione del muscolo cardiaco e che tale infiammazione è maggior-mente presente quanto più sono estese le aree cardia-che anomale.
Questo studio pone la sindrome di Brugada sotto una lu-ce completamente nuova:dimostra che non solo altera-zioni genetiche,ma anche un’infiammazione del cuore può causare la sindrome di Brugada; apre pertanto nuo-ve strade per l’identificazione dei pazienti con sindrome di Brugada ad alto rischio di morte improvvisa, i quali ne-cessitano dell’impianto di un defibrillatore.
La dott/ssa Vittoria Rizzello ha di recente pubblicato una interessante revisione sui meccanismi patogenetici della Sindrome di Brugada. In preatica quando 30 anni fa la sindrome fu descritta per la prima volta (1), fu etichettata come una canalopatia, geneticamente determina-ta, con cuore strutturalmente normale. Negli anni successivi alcune evidenze hanno documentato la presenza di alterazioni strutturali come infiltrazione (2) adiposa (simile alla displasia aritmogena del ventricolo destro), infiammazione (3,4) e fibrosi interstiziale (5). Queste alterazioni potrebbero avere delle implicazioni prognostiche in quanto possono costituire degli elementi di vulnerabilità aggiuntiva rispetto alla sola alterazione delle correnti del sodio e aumentare il rischio aritmico di questi pazienti.
Un interessante contributo alla comprensione dei meccanismi patogenetici della SdR viene fornito dal lavoro di Miles C e coll, pubblicato su JACC (6). Miles C, Asimaki A, Ster IC, et al. Biventricular Myocardial Fibrosis and Sudden Death in Patients With Brugada Syndrome. J Am Coll Cardiol. 2021;78:1511-21.
Gli autori hanno analizzato i cuori di 28 soggetti deceduti per morte cardiaca improvvisa di cui 6 con diagnosi ante-mortem dii Sindrome di Brugada, 18 con familiarità per morte improvvisa da Sindrome di Brugada e 4 con diagnosi di Sindrome di Brugada basata su test genetico (presenza di mutazioni patologiche/probabilmente patologiche del gene SCN5A). Come controlli sono stato utilizzati i cuori di soggetti morti improvvisamente per traumi non cardiaci e tossicità da oppioidi. L’estensione delle alterazioni strutturali a livello endocardico, epicardico e totale è stata valutata con un software automatico digitale (Visiopharm A/S) in grado di quantificare le diverse componenti tissutali. I prelievi di tessuto sono stati effettuati in 6 zone differenti, ossia il tratto di efflusso del ventricolo destro (TEVDx), la parete libera del VDx, il setto anteriore e inferiore, la parete anteriore e posteriore del ventricolo sinistro.
L’analisi morfometrica quantitativa automatica ha dimostrato che, nei deceduti con Sindrome di Brugada, nonostante la normalità dell’analisi istologica tradizionale, era presente un grado di fibrosi maggiore in tutte le sedi analizzate rispetto ai controlli (ratio 1.45; 95%IC: 1.22-1.71, P<0.001). La più alta percentuale di fibrosi era osservata nei campioni tissutali prelevati a livello epicardico del TEVdx (24%). I soggetti con Sindrome di Brugada ante-mortem presentavano il maggior grado di fibrosi (ratio 1.66; 95%IC: 1.25-2.20, P<0.001), seguiti dai soggetti con familiarità per Sindrome di Brugada (ratio 1.42; 95%IC: 1.17-1.71, P<0.001). Per contro, nei pazienti con sola evidenza genetica di mutazioni patologiche/ probabilmente patologiche del gene SCN5A non è stato osservato un grado di fibrosi maggiore rispetto ai controlli. L’analisi morfometrica non ha invece evidenziato differenze relative alla presenza di infiammazione o infiltrazione adiposa tra i deceduti per SdB e i controlli.
Considerazioni.
Lo studio di Miles C e coll appare rilevante ai fini della comprensione dei meccanismi fisiopatologici della Sindrome di Brugada in quanto conferma ed amplia, con la più numerosa coorte a oggi riportata di pazienti con Sindrome di Brugada deceduti per morte improvvisa, le precedenti osservazioni a favore di un substrato organico, in questo caso fibrotico, della sindrome. Tale substrato potrebbe avere un ruolo nel modulare l’induzione di aritmie in un setting elettrico, geneticamente determinato, di ridotta corrente di sodio. Infatti, in un precedente lavoro di Pieroni e coll (4) era stato evidenziato come, in pazienti con Sindrome di Brugada la presenza di bassi voltaggi bipolari al mapping elettroanatomico si associava alla presenza di infiltrati infiammatori alla biopsia mapping-guidata, con un aumento di 3 volte dell’inducibilità di aritmie allo studio elettrofisiologico nei pazienti con infiammazione rispetto ai pazienti senza infiamma-zione. Nello studio di Miles C e coll non sono stati individuati segni di infiammazione, tuttavia la fibrosi potrebbe rappresentare una fase evolutiva, più tardiva, di un processo infiammatorio pregresso, determinante, in maniera analoga, un’aumentata vulnerabilità aritmica. Un aspetto interessante dello studio di Miles C e coll è rappresentato dalla dimostrazione di aree di fibrosi anche a livello del ventricolo sinistro, indicando una possibile più diffusa estensione delle alterazioni elettriche della Sindrome di Brugada con la presenza di ulteriori circuiti di rientro anche al di fuori del Vdx. L’identificazione della fibrosi si configura quindi come un possibile target per la stratificazione in vivo del rischio aritmico nel paziente con sospetto o diagnosi certa di Sindrome di Brugada. Purtroppo, il delayed-enhancement alla RMN cardiaca, benchè utile nella quantificazione della fibrosi miocardica, ha dimostrato una scarsa sensibilità nell’identificazione della fibrosi interstiziale, tipica di questi paziente. Le nuove metodiche di T1-mapping e di quantificazione del volume extracellulare potrebbero essere più accurate in questo setting e pertanto studi dedicati sono fortemente auspicabili. Un dato sorprendente dello studio di Miles C e coll è quello riguardo l’assenza di una differente estensione della fibrosi nel gruppo dei soggetti con diagnosi di Sindrome di Brugada basata solo sull’identificazione di varianti patologiche del gene SCN5A, rispetto ai controlli. Gli autori attribuiscono tale risultato sia all’esiguità del gruppo (4 soggetti) che agli effetti pleiotropici, ancora ampiamente sconosciuti, delle mutazioni del gene SCN5A. Ciò indica come, nonostante questo studio abbia aggiunto un altro tassello importante, il puzzle della SdB sia ancora incompleto.
Referenze:
-Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992; 20: 1391-96
-Corrado D, Nava A, Buja G, et al. Familial cardiomyopathy underlies syndrome of right bundle branch block, ST segment elevation and sudden death. J Am Coll Cardiol. 1996;27:443-48 -Frustaci A, Priori SG, Pieroni M, et al. Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation. 2005;112:3680-87
-Pieroni M, Notarstefano P, Oliva A, et al. Electroanatomic and Pathologic Right Ventricular Outflow Tract Abnormalities in Patients With Brugada Syndrome. J Am Coll Cardiol. 2018;72:2747-57
-Nademanee K, Raju H, de Noronha SV, et al. Fibrosis, Connexin-43, and Conduction Abnormalities in the Brugada Syndrome. J Am Coll Cardiol. 2015;66:1976-86.
-Miles C, Asimaki A, Ster IC, et al. Biventricular Myocardial Fibrosis and Sudden Death in Patients With Brugada Syndrome. J Am Coll Cardiol. 2021;78:1511-21
INDICE ANALITICO





