 Cuore Sano
Cuore Sano
COME SI FORMANO LE
PLACCHE ATEROSCLEROTICHE?
======================================================
Sono state proposte numerose ipotesi per spiegare la formazione delle placche aterosclerotiche; tra queste le principali sono 2:
- l'ipotesi lipidica,
- l'ipotesi del danno endoteliale cronico.
1)L'ipotesi lipidica ipotizza che un aumento delle lipo-proteine a bassa densità del plasma (LDL-colestelolo) favorisca il loro ingresso nella parete arteriosa, con con-seguente accumulo di lipidi nelle cellule muscolari lisce e nei macrofagi,i quali si trasformano in cellule schiumose ("foam cells").
Tutti i lipidi, compreso il colesterolo non sono solubili in acqua, per cui per essere trasportati nel sangue,vengono veicolati da alcune proteine, chiamate apolipo-proteine. Il complesso proteine-lipidi costituisce le lipo-proteine (o lipoprotidi); comprendono una proteina e lipidi di varia natura:
- colesterolo,
- esteri del colesterolo,
- fosfolipidi,
- trigliceridi.
In sintesi le lipoproteine sono formate da una compo- nente proteica superficiale, che isola dall' ambiente plasmatico i lipidi, consentendone la loro solubilizzazione nel liquido che compone il sangue.
Le lipoproteine vengono distinte in base alla densità in lipoproteine più dense e più piccole (più proteina e meno grasso) e in lipoproteine meno dense e più grandi (più grasso che proteina).
Le lipoproteine in base alla densità possono essere:
- HDL (ad alta densità),
- LDL (a bassa densità),
- VLDL (a bassissima densità).
Le lipoproteine non sono sempre dannose; per esempio le HDL che trasportano il colesterolo verso il fegato per essere metabolizzato e trasformato, sono utili; al contrario le LDL che procedono verso le arterie, dove creano un danno perché depositano il colesterolo sulle loro pareti ostruendole, sono ovviamente nocive.
COMPITO DELLE LIPOPROTEINE E' QUELLO DI TRASPORTARE I GRASSI NELL' ORGANISMO.
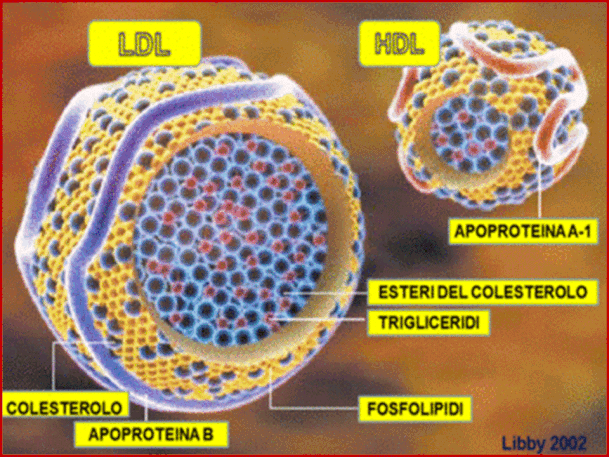
Di seguito sono riportate le principali composizioni di LDL e HDL.
Le LDL sono beta-lipoproteine costituite da:
-25% fosfolipidi,
-50% colesterolo,
-5% trgliceridi,
-20% proteine, di cui l' apoproteina B costituisce il 98%.
Le HDL sono alfa-lipoproteine costituite da:
-25% fosfolipidi,
-25% colesterolo,
-5% trigliceridi,
-45% proteine, di cui l' Apolipoproteina A costituisce il 90%.
Le LDL comprendono anche una frazione con le carat-teristiche di essere più piccole e dense; questa frazione di LDL ha una maggiore potenza aterogena e questo perchè ha una maggiore emivita plasmatica, dovuta ad una minore affinità per i recettori per le LDL.
Un aumento delle LDL piccole e dense è un reperto che caratterizza la sindrome plurimetabolica,la quale com-prende anche l'obesità centrale, l'insulino-resistenza,l' ipertrigliceridemia e bassi livelli di colesterolo HDL.
La sindrome plurimetabolica è la causa più frequente di aterosclerosi nella popolazione.
2) L'ipotesi della disfunzione endoteliale cronica vede in una lesione dell' endotelio, dovuta a diversi meccanismi, il primum movens del processo aterosclerotico.La disfun-zione endoteliale è uno stato patologico sistemico dell' endotelio caratterizzato da una riduzione della biodispo-nibilità di vasodilatatori, essenzialmente di ossido nitrico, portando a ridotta vasodilatazione endotelio-dipendente, nonché ad un dissesto nel metabolismo e funzione della parete vascolare. Il dissesto della funzione della parete consente ai grassi e al colesterolo di penetrare attra-verso queste lacerazioni e di depositarsi nella parete. Segue una adesione allo strato sotto-endoteliale di pia-strine, le quali si aggregano. Inoltre avviene una che-miotassi di monociti e linfociti T e rilascio di fattori di cre-scita derivati dalle piastrine e dai monociti, che favorisco-no la migrazione di cellule muscolari lisce dalla media verso l' intima dove si replicano e sintetizzano tessuto connettivo e proteoglicani formando una placca fibrosa.
Anche altre cellule come macrofagi, cellule endoteliali, etc producono fattori di crescita contribuendo così all' iperplasia di cellule muscolari lisce e alla produzione di matrice extracellulare. Quando l'endotelio diviene disfun-zionante, anche se le cellule endoteliali si dovessero ri-generare, non acquisisce mai una perfetta capacità fun-zionale.
Le due ipotesi patogenetiche non sono in antitesi tra di loro, ma si integrano.
I grassi del sangue ed in particolare il colesterolo sono trasportati nel sangue dalle lipoproteine a bassa densità (LDL). Esse non solo cedono continuativamente lipidi ai tessuti, ma sono sottoposte ad un numero elevatissimo di processi ossidativi. Quando le LDL divengono “LDL ossidate”, subiscono modificazioni che le rendono cito- tossiche nei confronti delle cellule endoteliali in quanto favoriscono l'aumento della permeabilità dell' endotelio vascolare ad alcuni elementi corpuscolati del sangue circolante come monociti e linfociti T (tipi particolari di globuli bianchi), i quali migrano così all' interno della pa-rete arteriosa. L’ endotelio risponde inoltre con una serie di eventi che scatenano una risposta infiammatoria. La risposta infiammatoria è implicata in tutte le fasi del pro-cesso aterosclerotico; in altri termini l' infiammazione ha un ruolo di primo piano per lo sviluppo di placca, per la sua progressione e per la sua vulnerabilità.
PERCHE' LE LIPOPROTEINE SI OSSIDANO?
L' ossidazione delle LDL è dovuta ad enzimi e metaboli-ti ossidanti prodotti dalle cellule della parete arteriosa, soprattutto dai monociti-macrofagi reclutati nell' intima in conseguenza del danno endoteliale a varia eziologia. Inizialmente si ha la perossidazione della com-ponente lipidica delle LDL, che interferisce scarsamente sull'in- terazione delle LDL con il recettore ApoB-E (o LDL-R); tali MM-LDL (LDL minimamente ossidate) sono “cavalli di Troia” (Hajjar:Journal of Biological Chemistry 1997, 272, 22975), fisicamente simili alle LDL, ma con un carico di macromolecole bioattive, che viene introdotto nella cel-lula con la endocitosi delle MM-LDL. Nelle fasi succes-sive, si generano prodotti dei lipidi perossidati e prodotti aldeidici (malondialdeide,MDA;4-idrossi-nonenale), che possono modificare covalentemente la componente pro-teica delle LDL; queste OX-LDL “sabotatori cellulari” non vengono più riconosciute da LDL-R, ma si legano agli "scavenger receptors" (SR: SR-A, CD36 e CD68). Poiché gli SR non sono soggetti a regolazione a feed-back-negativo, le OX-LDL non solo introducono nelle cellule che le fagocitano macromolecole attive, ma in ag-giunta causano l'accumulo intracellulare di esteri del colesterolo,responsabile della trasformazione in cellule schiumose o foam cells, caratteristiche del tessuto atero-sclerotico.
L'interazione con i corrispondenti recettori LDL-R e SR (e la conseguente generazione di messaggeri intracellulari, in particolare i radicali liberi dell'ossigeno o ROS) e l'introduzione nella cellula di prodotti ossidati sono la ba-se biochimica dell'azione patogena delle LDL. Le OX- LDL attivano nelle cellule (endotelio, macrofagi, cellule muscolari lisce), alcuni fattori di trascrizione (es. NF-κB), che inducono l'espressione di geni che codificano per molecole adesive, citochine e fattori di crescita e che danno l'avvio alla risposta infiammatoria.
Ad esempio, nell'endotelio, i geni per le molecole adesi-ve ICAM-1,VCAM-1 e E selectina,per il fattore chemiotat-tico MCP-1 e per il Fattore Tessutale sono sotto il con-trollo del fattore di trascrizione redox-sensibile NF-κB.
Kume N nel 1991 ha suggerito che le cellule endoteliali assorbono le OX-LDL attraverso una via recettoriale che non coinvolge gli scavenger receptors.
Sawamura T, Kume N, e altri nel 1997 hanno identificato il primo recettore delle cellule endoteliali per le Ox-LDL, che è stato denominato LOX-1 (lectinlike Ox-LDL recep-tor-1).
Studi sperimentali hanno dimostrato che le LDL ossidate possiedono numerose attività biologiche sulle cellule del-la parete arteriosa, inclusa un' azione citotossica diretta e un'azione mitogena sulle cellule muscolari lisce, sui ma-crofagi, sui fibroblasti e sulle cellule endoteliali. Nell'en-dotelio inducono l'espressione di molecole adesive per i leucociti; stimolano la produzione di sostanze chemiotat-tiche, che in parte rimangono legate alla superficie endoteliale e in parte sono liberate nel subendotelio; inol-tre favoriscono la sintesi di fattori di crescita per i mono-citi/macrofagi e per le cellule muscolari lisce; stimolano la sintesi di PAI-1 (plasminogen activator inhibitor-1) e di fattore tessutale, promuovendo la coagulazione; stimola-no la produzione di endotelina e inibiscono quella dell’ ossido nitrico (NO), inibendo la vasodilatazione endote-lio-dipendente. Sui macrofagi esercitano un effetto che-miotattico diretto; determinano la trasformazione in cel-lule schiumose; stimolano la produzione di citochine, fat-tori di crescita e metalloproteasi.Nelle cellule muscolari li-sce inducono la sintesi di MCP-1. Infine le LDL ossidate attivano le piastrine e ne provocano l'aggregazione.
La pericolosità delle LDL ossidate si estrinseca con un' azione citotossica per le cellule endoteliali, a cui conse- gue un richiamo di monociti e macrofagi e la stimola- zione alla proliferazione di cellule muscolari lisce. Con il passare del tempo queste cellule evolvono in macrofagi ed ingeriscono le LDL ossidate accumulando vacuoli lipi-dici nello spazio citoplasmatico formando così le cosid-dette "foam cells" (cellule schiumose). Le "foam cells" se-cernono sostanze infiammatorie e fattori di crescita che inducono una proliferazione di fibrocellule muscolari li-sce. Tale condizione porta alla formazione di una capsula fibrosa che ricopre l'accumulo di grasso. Queste placche fibro-lipidiche si formano durante un lungo periodo di tempo e caratterizzano la fase cosiddetta silente della aterosclerosi, la quale può durare anche alcuni decenni. L'evoluzione delle placche aterosclerotiche è fortemente influenzata dal contenuto in grassi e macrofagi, nel sen-so che un'elevata presenza di questi 2 elementi può fa-vorire la rottura delle placche stesse, alla quale rottura è legato lo scatenarsi degli eventi cardiovascolari (es. infarto del miocardio).

ROTTURA DEL CAPPUCCIO FIBROSO
Nel passato si credeva che la malattia aterosclerotica fosse un processo patologico progressivo, che portava progressivamente alla chiusura del vaso e quindi all’ evento acuto, anche fatale. Attualmente è ben noto la malattia progredisce a volte in modo brusco con im-provvise occlusioni dei vasi, che sono causate nella maggioranza dei casi da fenomeni di rottura della placca aterosclerotica con una conseguente trombosi occlusiva del vaso.Vi sono evidenze che anche placche di piccole dimensioni, che limitano solo parzialmente il flusso, pos-sono quindi essere sede di occlusioni arteriose acute.
È stato dimostrato che alcune caratteristiche strutturali i-dentificano le placche a maggiore rischio di rottura.
Esse comprendono:
1.un nucleo lipidico più grande,
2.un cappuccio fibroso superficiale più sottile,
3.la presenza di cellule infiammatorie attive nella placca,
4.una ridotta concentrazione di cellule muscolari lisce e di collageno nello strato fibroso superficiale,
5.un aumento dei vasa vasorum.
Anche la composizione del nucleo lipidico è un altro fattore determinante.Se sono presenti in maggioranza esteri del colesterolo, la consistenza della placca è sof-fice, mentre se vi è colesterolo cristallino la consistenza è più solida. Inoltre le placche che tendono a rompersi presentano più spesso un nucleo lipidico grande, eccen-trico e di consistenza soffice. In queste condizioni, le forze meccaniche sembrano concentrarsi sui suoi margi-ni della placca, proprio dove si verifica con maggior frequenza la rottura. Anche il cappuccio fibroso delle placche più tendenti alla rottura è più sottile ed è carat-terizzato da poche cellule muscolari lisce, da un basso contenuto di collagene e glicosaminoglicani e da un maggior numero di cellule infiammatorie quali macrofagi, linfociti T e mastociti.La presenza di cellule infiammato-rie giuoca un ruolo di primo piano nella fisiopatologia del-la rottura di placca. Uno dei meccanismi principali con cui favoriscono la rottura della placca è l’indebolimento del cappuccio fibroso, attraverso la secrezione di enzimi proteolitici quali le proteinasi e le metalloproteinasi.
Il concetto di instabilità o vulnerabilità di placca è stato proposto alla fine degli anni '80 per identifi-care le lesioni aterosclerotiche a rischio di altera-zione improvvisa in grado di provocare una trom-bosi. Il substrato anatomico della instabilità è sta-to identificato nella rottura o fissurazione della placca. Trattasi di una soluzione di continuità del cappuccio fibroso di un ateroma che è assai di frequente sottile.

L'esposizione del contenuto della placca al sangue cir-colante favorisce la cascata trombogenica. Usualmente la rottura interessa la zona di transizione tra la parte nor-male del vaso e il cappuccio fibroso, detta spalla della placca. Lo spessore del cappuccio in prossimità del sito di rottura è assai sottile, di frequente < 65 µm. Questo ti-po anatomico di placca viene definita "fibro-ateroma con cappuccio sottile". In un recente studio i pazienti diabetici hanno mostrato di avere con maggiore frequenza rispet-to ai non diabetici un cappuccio sottile. Il rischio di rottura di placca, e di conseguenza di un evento cardiova-sco-lare,è legato oltre che all'anatomia delle placche anche al loro stato infiammatorio. Infatti le placche anche se sono relativamente piccole ma se sono infiammate, sono con maggiore frequenza a rischio di rottura, rispetto alle plac-che di grandi dimensioni, ma non infiammate. Un indica-tore aspecifico del processo infiammatorio è la proteina C reattiva (PCR). A parità di valore di colesterolemia hanno un rischio maggiore, i pazienti con elevati valori di proteina C reattiva.Nella pratica i livelli di proteina C reat-tiva sono generalmente elevati nei pazienti che hanno subito un infarto del miocardio.
Poiché l'aumento della proteina C reattiva può essere le-gato a fenomeni infiammatori di varia natura come ad e-sempio anche una banale faringo-tonsillite, è ovvio che, ai fini della valutazione del rischio cardiovascolare, è ne-cessario effettuare la ricerca della proteina C reattiva in assenza di eventi infiammatori intercorrenti, ripetendola se necessario a breve distanza di tempo, qualora esi-stano dei dubbi.





